

Hopital Saint-Antoine - Bâtiment Kourilsky - 5ème étage - 184, rue du Faubourg Saint-Antoine, 75012 Paris, France



1. Nos centres d’intérêt
Notre équipe s’intéresse à la physiopathologie des troubles de la croissance impliquant le système des Insulin-like growth factors (IGFs). Le système des IGF est très impliqué dans le métabolisme, la prolifération, la différenciation, la survie cellulaires notamment lors du développement. IGF2 joue un rôle majeur dans le contrôle de la croissance fœtale et a la particularité d’être un gène soumis à l’empreinte parentale. L'empreinte génomique parentale est un processus épigénétique qui se traduit par l'expression mono-allélique de certains gènes en fonction de leur origine parentale. Cette expression est sous le contrôle de centre d’empreinte Imprinting Control Region, ICR, qui sont des régions différentiellement méthylées entre les deux allèles.
L’équipe travaille sur deux pathologies rares, en miroir clinique et moléculaire secondaires à la perturbation de l’empreinte parentale dans la région 11p15 et qui affectent la croissance fœtale : le syndrome de Silver-Russell (SRS, retard de croissance) et le syndrome de Beckwith-Wiedmann (BWS, croissance excessive avec un risque accru de développement de tumeurs embryonnaires). Nous avons également rapporté la première cohorte française de patients atteints du syndrome de Temple (TS14), une autre maladie liée à l’empreinte, responsable d'un retard de croissance et de troubles métaboliques et endocriniens précoces, secondaire à des anomalies moléculaires de la région 14q32.
Ces syndromes sont secondaires à des anomalies génétiques ou épigénétiques (en particulier des anomalies de méthylation au niveau des centres d’empreinte) aboutissant à des défauts de l’expression des gènes soumis à empreinte parentale. L'objectif principal de notre équipe est d'améliorer les connaissances sur les mécanismes physiopathologiques et environnementaux impliqués dans la régulation de la croissance fœtale et postnatale. Pour ce faire, nous bénéficions d’une approche transversale par des études cliniques et fondamentales à partir de cohortes.
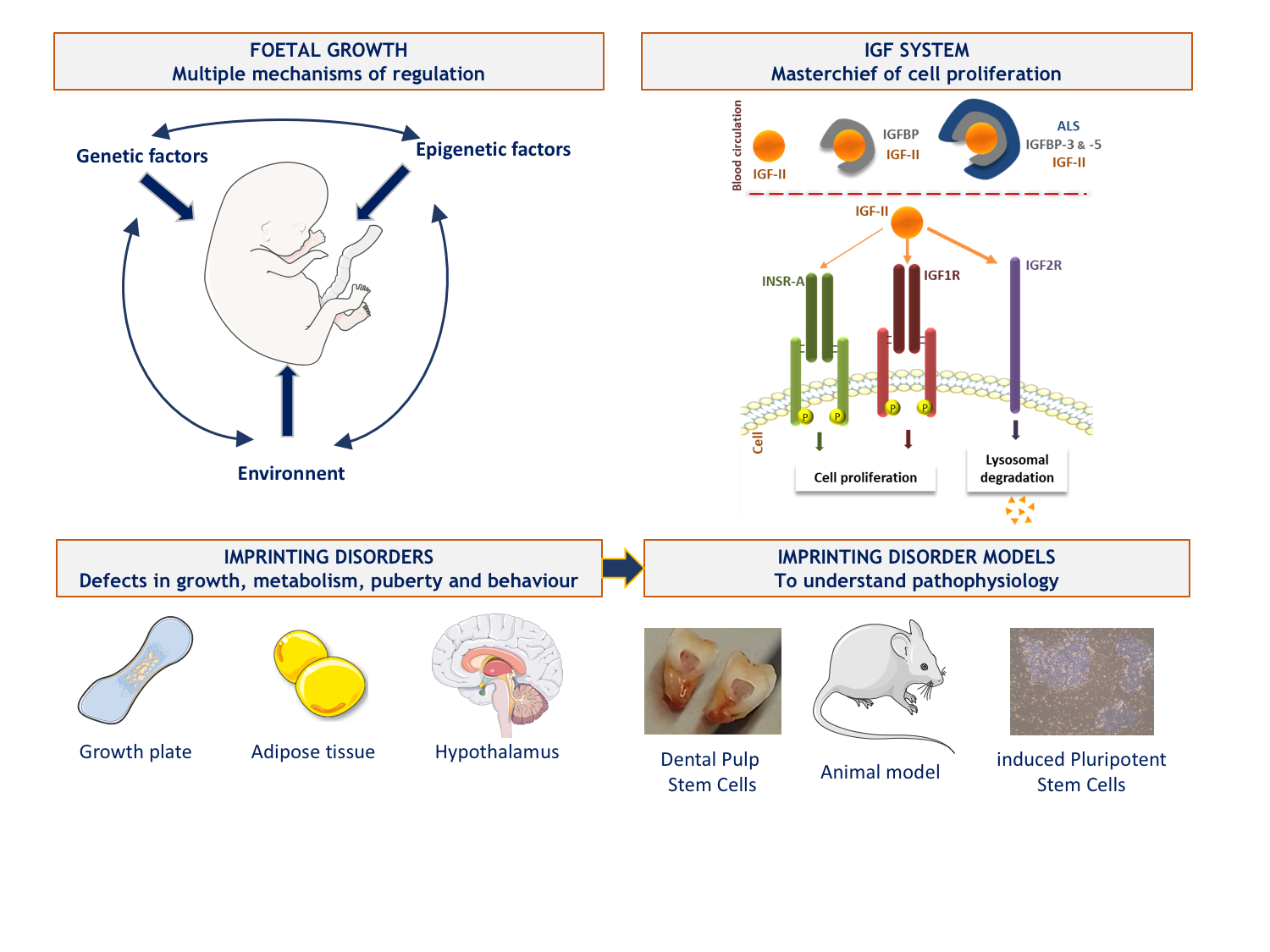
2. Les travaux récents et en cours
A/ Nous avons mis au point un panel de séquençage haut débit de gènes impliqués dans la restriction ou l’excès de croissance fœtale afin d'améliorer le diagnostic des anomalies génétiques chez les patients présentant une anomalie de la croissance fœtale en France. En dehors des gènes déjà décrits dans des syndromes proches du SRS et du BWS, nous avons inclus certains gènes d’intérêt pour lesquels aucune anomalie n'a été identifiée chez l'être humain mais qui sont théoriquement impliqués dans la régulation de la croissance fœtale. La plupart d'entre eux font partie du système des IGF comme leurs protéines de liaison ou les effecteurs en aval de leur récepteur commun, IRS1 et IRS2.
B/ En absence de modèle d’étude tout à fait pertinent pour étudier la physiopathologie des maladies d’empreinte auxquelles nous nous intéressons, nous avons développé deux modèles cellulaires principaux.
Nous disposons ainsi d’un modèle prometteur à partir de cellules souches de la pulpe dentaire - que nous pouvons collecter lorsque les patients ont des soins d’extraction dentaire programmée - dont la multipotence permet la différenciation en cellules d’intérêt, impliquées dans le phénotype des patients. Ce travail très récent a été réalisé en parallèle avec la reprogrammation des cellules souches pluripotentes induites (iPSC) à partir des leucocytes. Nous avons pu nous affranchir du profil d'hyperméthylation identifié pour la quasi-totalité des ICR dans ces cellules - comme d'autres équipes l'avaient également rapporté – en mettant au point des conditions de culture de reprogrammation permettant de restaurer une méthylation équilibrée dans certains clones. Ce protocole innovant a fait l'objet d'une publication et d’une demande de brevet (n°22306056.7).
Actuellement, nous différencions ces deux types de cellules souches en cellules impliquées dans le phénotype des patients telles que les chondrocytes hypertrophiques (cellules clés de la plaque de croissance chez l'homme) ou les adipocytes. Ainsi, nous pourrons caractériser les différences transcriptomiques, développementales et fonctionnelles existant dans ces cellules issues de contrôles et de patients et mieux comprendre les conséquences physiopathologiques des maladies d’empreinte, premier pas vers la mise en évidence et le développement de nouvelles cibles thérapeutiques.
Par ailleurs, à l’avenir, nous envisageons de compléter les données issues de ces études cellulaires par des études sur des modèles animaux murins de perturbation de l’empreinte parentale et du système des IGF grâce au développement des techniques d’édition du génome et de l’épigénome.
C/ D’autre part, chez les patients avec maladies d’empreinte, les défauts de méthylation peuvent s'étendre à d'autres loci soumis à empreinte en dehors de la région initialement identifiée comme perturbée. Ce phénomène est appelé "atteinte de l'empreinte multilocus" (multilocus imprinting disorder, MLID). Aujourd'hui, la prévalence et les conséquences cliniques de la MLID sont mal décrites. Avec l'aide des technologies de séquençage haut débit, nous développons de nouveaux outils moléculaires permettant d’analyser la méthylation d’un grand nombre de loci soumis à empreinte en un seul temps.
D/ Enfin, notre dernier axe de recherche consiste à étudier l'impact de l'environnement sur les maladies d'empreinte avec anomalie de la croissance fœtale. Les difficultés rencontrées pour obtenir des iPSCs avec une méthylation normale au niveau des ICRs l’influence des conditions de culture font échos à la prévalence accrue des maladies de l'empreinte chez les enfants nés par les technologies de procréation assistée, un fait déjà documenté dans la littérature et précédemment étudié dans notre équipe par un PHRC (Pr Yves Le Bouc). En effet, l’origine des anomalies épigénétiques responsables de la majorité des maladies liées à l’empreinte que nous étudions n’est pas connue mais les phases critiques de mise en place et maintien de la méthylation des ICR se situent lors de la gamétogénèse et dans les premiers jours post-conceptionnels. Les évènements environnementaux survenant à ces périodes-là sont donc intéressant à étudier pour évaluer leur effet sur la régulation de l’empreinte parentale.
Irène Netchine : ORCID 0000-0003-1324-3389
AFIF-SSR-PAG: https://silver-russell.fr/
RADICO-IDMET’ cohort for patients with Imprinting Disorders
https://www.radico.fr/fr/connaitre-radico/nos-cohortes-et-autres-programmes-associes/80-radico/146-radico-idmet
Marie-Laure Sobrier: ORCID ID 0000-0001-6396-100X
Eloïse Giabicani : ORCID ID 0000-0001-5360-8616
https://fr.linkedin.com/in/elo%C3%AFse-giabicani-099924153
Frédéric Brioude : ORCID 0000-0001-8122-760X
Laurent Kappeler : ORCID 0000-0001-7971-0838
https://www.researchgate.net/profile/Laurent-Kappeler-2
Clémence Girardet: ORCID 0000-0002-0302-3350
https://www.researchgate.net/profile/Girardet-Clemence
International guideline on genetic testing of children with short stature.
Dauber A, Jorge AAL, Nilsson O, Dekkers OM, Argente J, Netchine I, Backeljauw P, Baron J, Bertola DR, Clayton P, Davies JH, Edouard T, Eggermann T, Gevers EF, Grigelioniene G, Heath KE, Hee Jee Y, Lapunzina P, Mortier G, Pruhova S, Storr HL, Wakeling E, Ferreira CR, Hasegawa T, Hokken-Koelega A, Linglart A, Luo X, Wang X, Hwa V, Gregory LC, Buonocore F, Dattani M, Cianfarani S, Wit JM. Eur J Endocrinol. 2026 Jan 16:lvag013. doi: 10.1093/ejendo/lvag013.
(lien hypertexte: https://academic.oup.com/ejendo/advance-article/doi/10.1093/ejendo/lvag013/8427429?login=true)
EndoCompass Project: Research Roadmap for Growth Disorders.
Gevers EF, Hokken-Koelega AC, Tauber M, Binder G, Bochukova EG, Bouret SG, Caixàs A, Davies JH, Dauber A, Edouard T, Eggermann T, Giabicani E, Netchine I, Nilsson O, Saravinovska K, van der Steen M, Tartaglia M, Tatton-Brown K, Temple IK, Yart A, Zenker M. Horm Res Paediatr. 2025;98(Suppl. 2):91-104. doi: 10.1159/000549204.
(lien hypertexte: https://karger.com/hrp/article/98/Suppl.%202/91/939254/EndoCompass-Project-Research-Roadmap-for-Growth)
Hypomethylation of the MEG8:Int2-DMR in patients with pathogenic PLAG1 variants suggests new role of the chr14q32 imprinting cluster in Silver-Russell syndrome.
D'Angelo E, Pignata L, Cecere F, Vimercati A, Cubellis MV, Saadat A, Giaccari C, Thibaud N, Eggermann T, Fernández-Fructuoso JR, Russo S, Netchine I, Cerrato F, Riccio A, Brioude F. Clin Epigenetics. 2025 Nov 23;18(1):2. doi: 10.1186/s13148-025-02024-6.
(lien hypertexte: https://link.springer.com/article/10.1186/s13148-025-02024-6)
EndoCompass project: research roadmap for growth disorders.
Gevers EF, Hokken-Koelega AC, Tauber M, Binder G, Bochukova EG, Bouret SG, Caixàs A, Davies JH, Dauber A, Edouard T, Eggermann T, Giabicani E, Netchine I, Nilsson O, Saravinovska K, van der Steen M, Tartaglia M, Tatton-Brown K, Temple IK, Yart A, Zenker M. Eur J Endocrinol. 2025 Oct 17;193(Supplement_2):ii72-ii83. doi: 10.1093/ejendo/lvaf070.
(lien hypertexte: https://academic.oup.com/ejendo/article/193/Supplement_2/ii72/8281949?login=true)
Advances in congenital adrenal hyperplasia newborn screening: 11-ketotestosterone and 21-deoxycortisone as additional discriminatory biomarkers.
Sow C, Nguyen-Khoa T, Moreau C, Fiet J, Bachelot G, Ribault B, Chaussenery O, Al Zarga L, Desmons A, Brioude F, Kariyawasam D, Stoupa A, Polak M, Houang M, Lamazière A; FIRENDO Group. Eur J Endocrinol. 2025 Nov 26;193(6):677-686. doi: 10.1093/ejendo/lvaf208.
(lien hypertexte : https://academic.oup.com/ejendo/article/193/6/677/8280089?login=true)
Early Growth Hormone Treatment Enhances Growth and Nutritional Status in Silver-Russell Syndrome.
Giabicani E, Billette de Villemeur R, Acher M, Cochet M, Perrière A, Dubern B, Netchine I. J Clin Endocrinol Metab. 2025 Aug 19:dgaf452. doi: 10.1210/clinem/dgaf452.
(lien hypertexte : https://academic.oup.com/jcem/advance-article-abstract/doi/10.1210/clinem/dgaf452/8237732?redirectedFrom=fulltext&login=true)
ImprintCap, a powerful NGS-based technology to investigate the molecular background of imprinting disorders.
Brioude F, Haagmans MA, Mannens M, Netchine I, Alders M, Henneman P, Bliek J. Clin Epigenetics. 2025 Jul 7;17(1):119. doi: 10.1186/s13148-025-01916-x.
(lien hypertexte : https://link.springer.com/article/10.1186/s13148-025-01916-x)
The RaDiCo information system for rare disease cohorts
Landais P, Gueguen S, Clement A, Amselem S; RaDiCo Program.. Orphanet J Rare Dis. 2025 Apr 8;20(1):166. doi: 10.1186/s13023-025-03629-z.
((lien hypertexte : https://link.springer.com/article/10.1186/s13023-025-03629-z)
Silver-Russell Syndrome.
Saal HM, Harbison MD, Netchine I. 2002 Nov 2 [updated 2025 Jan 9]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2026.
((lien hypertexte : https://www.ncbi.nlm.nih.gov/books/NBK1324/)
The RNA-binding protein TRIM71 is essential for hearing in humans and mice and times auditory sensory organ development.
Li XJ, Morgan C, Duy PQ, Evsen L, Hao LT, Machavoine R, Belhous K, Ernest S, Denoyelle F, Mignot C, Brioude F, Parodi M, Li L, Huang H, Nadar Ponniah PT, Kolanus W, Kahle KT, Marlin S, Doetzlhofer A. Proc Natl Acad Sci U S A. 2025 Sep 9;122(36):e2505811122. doi: 10.1073/pnas.2505811122.
(lien hypertexte : https://www.pnas.org/doi/10.1073/pnas.2505811122?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0pubmed)
A recurrent splice variant sheds light on 11beta-hydroxylase deficiency in a unique large cohort.
Janot C, Mallet D, Janin A, Bertherat J, Brauner R, Brioude F, Cartault A, Daval-Cote M, Espiard S, Houang M, Kraus Friedmann J, Lefebvre H, Martinerie L, Mayer A, Mazoyer H, Menassa R, Morel Y, Pienkowski C, Ribault V, Plotton I, Teoli J, Brac de La Perrière A, Roucher-Boulez F. J Clin Endocrinol Metab. 2025 Aug 19:dgaf468. doi: 10.1210/clinem/dgaf468.
(lien hypertexte : https://academic.oup.com/jcem/advance-article-abstract/doi/10.1210/clinem/dgaf468/8237788?redirectedFrom=fulltext&login=true)
Silver-Russell Syndrome in 2025: Is It Still a Distinct Diagnostic Entity?
Giabicani E, Perriere A, Netchine I. J Clin Endocrinol Metab. 2025 Oct 16;110(11):e3905-e3906. doi: 10.1210/clinem/dgae902.
(lien hypertexte : https://academic.oup.com/jcem/article/110/11/e3905/7941443?login=true)
Multi-locus imprinting disturbance (MLID): interim joint statement for clinical and molecular diagnosis.
Mackay DJG, Gazdagh G, Monk D, Brioude F, Giabicani E, Krzyzewska IM, Kalish JM, Maas SM, Kagami M, Beygo J, Kahre T, Tenorio-Castano J, Ambrozaitytė L, Burnytė B, Cerrato F, Davies JH, Ferrero GB, Fjodorova O, Manero-Azua A, Pereda A, Russo S, Tannorella P, Temple KI, Õunap K, Riccio A, de Nanclares GP, Maher ER, Lapunzina P, Netchine I, Eggermann T, Bliek J, Tümer Z. Clin Epigenetics. 2024 Aug 1;16(1):99. doi: 10.1186/s13148-024-01713-y.
((lien hypertexte : https://link.springer.com/article/10.1186/s13148-024-01713-y)
Perinatal features of children with Silver-Russell syndrome due to 11p15 loss of methylation.
Darneau D, Giabicani E, Netchine I, Pham A. Front Pediatr. 2024 Apr 4;12:1367433. doi: 10.3389/fped.2024.1367433.
Pubertal origin of growth retardation in inborn errors of protein metabolism: A longitudinal cohort study.
Busiah K, Roda C, Crosnier AS, Brassier A, Servais A, Wicker C, Dubois S, Assoun M, Belloche C, Ottolenghi C, Pontoizeau C, Souberbielle JC, Piketty ML, Perin L, Le Bouc Y, Arnoux JB, Netchine I, Imbard A, de Lonlay P. Mol Genet Metab. 2024 Mar;141(3):108123. doi: 10.1016/j.ymgme.2023.108123.
Ciliopathy due to POC1A deficiency: clinical and metabolic features, and cellular modeling.
Perge K, Capel E, Villanueva C, Gautheron J, Diallo S, Auclair M, Rondeau S, Morichon R, Brioude F, Jéru I, Rossi M, Nicolino M, Vigouroux C. Eur J Endocrinol. 2024 Feb 1;190(2):151-164. doi: 10.1093/ejendo/lvae009.
Post-ligation cardiac syndrome after surgical versus transcatheter closure of patent ductus arteriosus in low body weight premature infants: a multicenter retrospective cohort study.
Duboue PM, Padovani P, Bouteiller XP, Martin-Kabore F, Benbrik N, Gronier CG, Bouissou A, Garnier E, Mitanchez D, Flamant C, Rozé JC, Baruteau AE, Lefort B. Eur J Pediatr. 2024 May;183(5):2193-2201. doi: 10.1007/s00431-024-05481-y.
Electronic reporting of rare endocrine conditions within a clinical network: results from the EuRRECa project.
Ali SR, Bryce J, Priego-Zurita AL, Cherenko M, Smythe C, de Rooij TM, Cools M, Danne T, Katugampola H, Dekkers OM, Hiort O, Linglart A, Netchine I, Nordenstrom A, Attila P, Persani L, Reisch N, Smyth A, Sumnik Z, Taruscio D, Visser WE, Pereira AM, Appelman-Dijkstra NM, Ahmed SF. Endocr Connect. 2023 Nov 20;12(12):e230434. doi: 10.1530/EC-23-0434.
Executive functioning in adolescents and adults with Silver-Russell syndrome.
Burgevin M, Lacroix A, Ollivier F, Bourdet K, Coutant R, Donadille B, Faivre L, Manouvrier-Hanu S, Petit F, Thauvin-Robinet C, Toutain A, Netchine I, Odent S. PLoS One. 2023 Jan 20;18(1):e0279745. doi: 10.1371/journal.pone.0279745.
Imprinting disorders.
Eggermann T, Monk D, de Nanclares GP, Kagami M, Giabicani E, Riccio A, Tümer Z, Kalish JM, Tauber M, Duis J, Weksberg R, Maher ER, Begemann M, Elbracht M. Nat Rev Dis Primers. 2023 Jun 29;9(1):33. doi: 10.1038/s41572-023-00443-4.
Maintenance of methylation profile in imprinting control regions in human induced pluripotent stem cells.
Pham A, Selenou C, Giabicani E, Fontaine V, Marteau S, Brioude F, David L, Mitanchez D, Sobrier ML, Netchine I. Clin Epigenetics. 2022 14(1):190. doi: 10.1186/s13148-022-01410-8.
Dental pulp stem cells as a promising model to study imprinting diseases.
Giabicani E, Pham A, Sélénou C, Sobrier ML, Andrique C, Lesieur J, LinglartA, Poliard A, Chaussain C, Netchine I. Int J Oral Sci. 2022 Apr 2;14(1):19. doi:10.1038/s41368-022-00169-1.
IGF2 : Development, Genetic and Epigenetic Abnormalities.
Sélénou C, Brioude F, Giabicani E, Sobrier ML, Netchine I. Cells. 2022 11(12):1886. doi: 10.3390/cells11121886.
Low Maternal DLK1 Levels at 26 Weeks Is Associated With Small for Gestational Age at Birth.
Pham A, Mitanchez D, Forhan A, Perin L, Le Bouc Y, Brioude F, Sobrier ML,Heude B, Netchine I. Front Endocrinol (Lausanne). 2022 13:836731. doi: 10.3389/fendo.2022.836731.
Increasing knowledge in IGF1R defects: lessons from 35 new patients.
Giabicani E, Willems M, Steunou V, Chantot-Bastaraud S, Thibaud N, Abi Habib W, Azzi S, Lam B, Bérard L, Bony-Trifunovic H, Brachet C, Brischoux-Boucher E, Caldagues E, Coutant R, Cuvelier ML, Gelwane G, Guemas I, Houang M, Isidor B, Jeandel C, Lespinasse J, Naud-Saudreau C, Jesuran-Perelroizen M, Perrin L, Piard J, Sechter C, Souchon PF, Storey C, Thomas D, Le Bouc Y, Rossignol S, Netchine I, Brioude F. J Med Genet. 2020 57(3):160-168. doi:10.1136/jmedgenet-2019-106328.
Overgrowth syndromes - clinical and molecular aspects and tumour risk.
Brioude F, Toutain A, Giabicani E, Cottereau E, Cormier-Daire V, Netchine I. Nat Rev Endocrinol. 2019 15(5):299-311. doi: 10.1038/s41574-019-0180-z.
Transcriptional profiling at the DLK1/MEG3 domain explains clinical overlap between imprinting disorders.
Abi Habib W, Brioude F, Azzi S, Rossignol S, Linglart A, Sobrier ML, Giabicani É, Steunou V, Harbison MD, Le Bouc Y, Netchine I. Sci Adv. 2019 5(2):eaau9425. doi:10.1126/sciadv.aau9425.
Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction.
Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F,Salem J, Yuen T, Azzi S, Le Bouc Y, Harbison MD, Netchine I. Genet Med. 2018 20(2):250-258. doi: 10.1038/gim.2017.105
Chromosome 14q32.2 Imprinted Region Disruption as an Alternative Molecular Diagnosis of Silver-Russell Syndrome.
Geoffron S, Abi Habib W, Chantot-Bastaraud S, Dubern B, Steunou V, Azzi S, Afenjar A, Busa T, Pinheiro Canton A, Chalouhi C, Dufourg MN, Esteva B, Fradin M, Geneviève D, Heide S, Isidor B, Linglart A, Morice Picard F, Naud-Saudreau C, Oliver Petit I, Philip N, Pienkowski C, Rio M, Rossignol S, Tauber M, Thevenon J, Vu-Hong TA, Harbison MD, Salem J, Brioude F, Netchine I, Giabicani E. J Clin Endocrinol Metab. 2018 103(7):2436-2446. doi: 10.1210/jc.2017-02152. PMID: 29659920
Diagnosis and management of Silver-Russell syndrome: first international consensus statement.
Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J,Canton AP, Chrzanowska KH, Davies JH, Dias RP, Dubern B, Elbracht M, Giabicani E, Grimberg A, Grønskov K, Hokken-Koelega AC, Jorge AA, Kagami M, Linglart A,Maghnie M, Mohnike K, Monk D, Moore GE, Murray PG, Ogata T, Petit IO, Russo S,Said E, Toumba M, Tümer Z, Binder G, Eggermann T, Harbison MD, Temple IK, MackayDJ, Netchine I. Nat Rev Endocrinol. 2017 13(2):105-124. doi: 10.1038/nrendo.2016.138.
Bâtiment Kourilsky
34 rue Crozatier
75012 PARIS
Sorbonne Université
27 rue Chaligny
75012 PARIS













